小鼠的Memo1缺失导致过早衰老和代谢不平衡,部分类似于Fgf23和Klotho功能丧失动物。我们报道了Memo的氧化还原功能在出生后诱导的全身Memo缺乏症(cKO)小鼠肾脏FGF23-Klotho信号传导中的作用。Memo cKO小鼠显示fgf23驱动的肾脏ERK磷酸化和转录反应受损。FGF23的作用涉及肾脏氧化敏感蛋白磷酸酪氨酸磷酸酶的激活。氧化还原蛋白质组学发现Memo cKO中Rho-GDP解离抑制剂1 (Rho-GDI1)硫醇含量过高,我们检测到Memo的氧化还原功能与Rho-GDI1 Cys79的氧化之间存在功能相互作用。在分离的细胞系统中,Rho-GDI1不直接影响fgf23驱动的细胞信号传导,但我们在Memo cKO小鼠的肾脏中检测到Rho-GDI1依赖性小Rho-GTPase蛋白的丰度和活性受到干扰。总的来说,本研究揭示了肾脏FGF23信号调控中以前未知的层面,并将Memo与小Rho-GTPases网络联系起来。
成纤维细胞生长因子(FGF) 23在慢性肾脏疾病(CKD)的进展中起关键作用。FGF23浓度升高与有或无CKD患者的血管钙化、心脏病和死亡率等并发症相关(Marthi et al. 2018)。FGF23由成骨细胞或骨细胞在维生素D、甲状旁腺激素、细胞外磷酸盐或炎症细胞因子等刺激下分泌(Richter and Faul 2018)。FGF23利用Klotho作为辅助受体激活FGFR1的典型通路,刺激甲状旁腺或肾脏的ERK信号传导(Urakawa et al. 2006)。CKD实验模型中异常的肾脏fgf23驱动的信号传导具有有害的细胞后果,如诱导炎症反应(Dai等人,2012),FGFR2介导的白细胞募集受损(Rossaint等人,2016)和肾脏纤维化(Smith等人,2017;Hao et al. 2021)。然而,潜在的分子机制尚不完全清楚。
细胞运动介质1 (Memo)是一种进化上保守的蛋白质,与细胞骨架和受体酪氨酸激酶相关(Marone et al. 2004;Zaoui et al. 2008;Jiang et al. 2013;Schotanus and Van Otterloo 2020)。Memo在结构上类似于细菌的非血红素铁双加氧酶(Qiu et al. 2008),具有还原铜的氧化还原功能。哺乳动物细胞突起中烟酰胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶(NOX)的活性需要备忘录(MacDonald et al. 2014)。在小鼠中,Memo1缺失导致胚胎死亡。使用Cre-lox技术,自然诱导的Memo1外显子2 (Memo cKO)普遍缺失导致小鼠过早衰老,伴有侏儒症、脱发、头发变白、皮下脂肪减少、性腺功能减退和步态异常(Haenzi et al. 2014)。此外,与对照组相比,这些动物对胰岛素过敏,葡萄糖耐量增加(Haenzi et al. 2014)。这些发现在早衰模型中很少看到,并且与广泛研究的Fgf23和klotho缺陷小鼠模型惊人地相似(Kuro-o et al. 1997;Shimada et al. 2004)。此外,在进行Memo1外显子2切除重组后4至8周,Memo cKO动物出现肾衰竭,血清FGF23和钙浓度升高,以及骨和矿物质疾病(Haenzi et al. 2014;Moor et al. 2018,2020)。这与Fgf23和klotho缺陷菌株部分重叠(Kuro-o et al. 1997;Shimada et al. 2004)。然而,Fgf23和Klotho功能丧失菌株的高磷血症(Kuro-o et al. 1997;Shimada et al. 2004)在Memo cKO小鼠中未发现。然而,Memo cKO小鼠也表现出肾表皮生长因子表达增加,镁转运蛋白水平和血清浓度增加(Haenzi et al. 2014;Moor et al. 2020),它们与远端肾元的功能密切相关(Groenestege et al. 2007)。表皮生长因子信号传导促进肾脏磷酸盐排泄,这可能解释了Memo cKO中正常的磷尿(Arar et al. 1995,1999)。
然而,细胞培养实验的结果支持了FGF23、FGFR共受体klotho和Memo在一个共同的信号通路中起作用的假设:首先,Memo与FGFR1和FRS2以及在配体结合后被募集到FGFR的接蛋白共同免疫沉淀。其次,细胞中的Memo敲低降低了FGF2或FGF23处理诱导的FRS2磷酸化(Haenzi et al. 2014)。
这些发现是否适用于复杂的器官,如肾脏(FGF23的主要作用部位),目前尚不清楚。此外,Memo影响RTK信号的机制不仅仅是作为接头蛋白支架的潜在作用(Marone et al. 2004;Haenzi et al. 2014;Newkirk et al. 2018)不完全理解。
在这里,我们假设Memo通过其氧化还原酶的功能调节肾fgf23诱导的细胞信号传导。在这里提出的实验中,我们证明Memo在肾脏中fgf23驱动的信号传导中起关键作用。我们发现Memo调节小rho - gtpase的活性和丰度,以及伴随FGF23细胞内信号反应的蛋白半胱氨酸氧化。
小鼠喂食标准饲料(TS3242 Kliba Nafnag, Kaiseraugst, Switzerland),并按12/12或14/10光暗循环饲养。经过10代回交到C57BL/ 6j背景的小鼠(Haenzi et al. 2014)与携带由β -肌动蛋白启动子/增强子控制的他莫昔芬诱导的Cre重组酶的CreERTM转基因小鼠(Hayashi and McMahon 2002)杂交。采用引物对耳部穿刺活检DNA进行PCR,确定基因型:Memo正向5′-CCCTCTCATCTGGCTTGGTA-3′,Memo反向5′-GCTGCATATGCTCACAAAGG-3′,Cre正向5′-AGGTTCGTGCACTCATGGA-3′,Cre反向5′-TCACCAGTTTAGTTACCC-3′。在目前的研究中,除了图5中报道的每种基因型的性别比例为50:50的实验外,所有使用的动物都是雄性。4周龄时,每天3次腹腔注射2mg他莫昔芬(T5648 Sigma-Aldrich,通过瑞士默克公司分发)诱导Memo丧失。没有Cre但用他莫昔芬治疗的母窝作为对照。对于FGF23, 6.5周龄小鼠饥饿6小时后,腹腔注射220 ng/g体重的重组小鼠FGF23 (2629-FG-025 R&D Systems, Minneapolis, MN, USA),以PBS-BSA 0.1%或PBS-BSA 0.1%载药,注射量为4μL/g体重。1小时后解剖小鼠,该方案改编自(Gattineni et al. 2014)。通过抛硬币的方式随机分配治疗方案。动物在麻醉状态下,1 h后以终末放血方式安乐死。对于调节FGF23信号的指示性预处理,在注射FGF23或PBS-BSA 0.1%前1小时,动物额外接受20 mg/kg (13721-39-6 Sigma-Aldrich) (Wang et al. 2013)或0.9%氯化钠的腹腔注射。
从ATCC获得HEK293细胞,使用LookOut?pcr检测试剂盒(MP0035-1KT Sigma-Aldrich)检测支原体阴性。HEK293-Klotho细胞稳定表达Klotho是Bettina Lorenz-Depiereux, HelmholtzZentrum m
Hek293-Klotho细胞以20万细胞/孔的密度接种在12孔组织培养板上,用ARHGDIA Ambion?Silencer?Select Pre-Designed siRNA (4427038 Ambion,通过瑞士Reinach的Thermo Fisher Scientific公司分发)或RhoGDI1 (ARHGDIA) Human siRNA Oligo Duplex(位点ID 396) (SR300287 OriGene Technologies Inc., Rockville MD, USA)使用Lipofectamine 3000转染试剂(L3000015 Invitrogen,通过Thermo Fisher Scientific公司分发)转染。Reinach,瑞士),遵循制造商的协议。转染Rho-GDI1或阴性对照sirna 72 h后,用载药(0.1% BSA)或100 ng/ml FGF23 (SRP3039 Sigma-Aldrich, Buchs, Switzerland)处理细胞5或15分钟。处理后,在摇床上孵育1小时,在RIPA (R0278 Sigma-Aldrich)中提取细胞蛋白,然后在10000 g离心。
每个样本取2只小鼠的一半肾脏,分别用10 mM Tris-HCl、pH 7.2、250 mM蔗糖、1 mM EDTA、150 mM KCl和1 mM PMSF匀浆,然后纺丝。蛋白硫醇和羰基分别用0.2 mM的5′-碘乙酰氨基荧光素(IAF)或1 mM的荧光素-5-硫代氨基脲(FTSC)标记,在37°C黑暗中孵育150分钟。用20% TCA沉淀蛋白,4°C, 20000 × g离心3min。重悬蛋白球,分别用100%乙醇/乙酸乙酯(1:1)和96%丙酮洗涤羰基和硫基。将微球用Tris-HCl 0.5 M pH 6.8,甘油10%,SDS 0.5%和溴酚蓝重悬,应用于SDS- page。凝胶在Typhoon Trio Scanner 9400 (Control v5.0 +可变模式成成仪- ra 501: PRT < I/06/004, GE Healthcare, buckinghamshire, UK)上进行荧光扫描;激发,490-495 nm;发射光谱,515-520 nm)。使用Quantity One图像分析软件(BioRad, Hercules, CA, USA)分析蛋白相关荧光强度(任意单位,AU)。凝胶用胶体考马斯亮蓝G250染色并扫描。
在凝胶电泳(2DGE)的二维分析中,首先用pI(第一维:等电聚焦IEF)分离蛋白质,然后根据分子量进行正交分离(第二维:SDS-PAGE)。蛋白质在5 M尿素、2 M硫脲、2% CHAPS、4%两性溶液(Pharmalyte 3 - 10, amerham - pharmacia Biotech, Little Chalfont, Bucks, UK)、1% Destreak试剂(amerham - pharmacia Biotech, Buckinghamshire, UK)和微量溴酚蓝中再水化,然后固定在7 cm IPG条(pH 3 - 10: 70 × 3 × 0.5 mm)和线性梯度(NL)中(GE Healthcare Immobiline?Dry Strip IPG, GE17-6001-11 biosciences AB, Bio-Rad, Hercules, CA, USA)。蛋白质在Protean IEF Cell (Bio-Rad)中室温聚焦至少15小时,按照以下步骤:(1)线性增加电压至250 V,持续15分钟,(2)10,000 V,持续2小时(50μa /条),(3)在20,000 V聚焦,(4)保持在500 V。IEF后,试纸在平衡缓冲液(6 M尿素,0.375 M Tris, pH 8.8, 2% SDS, 20%甘油,含2% DTT)中平衡20分钟,然后在含2.5%碘乙酰胺的平衡缓冲液中平衡20分钟。将IPG条带加载到12% SDS-PAGE凝胶(PROTEAN Plus Dodeca Cell, BioRad)上。如上所述,对凝胶进行荧光扫描,用考马斯蓝染色,然后进行密度测定。Progenesis SameSpots软件(S/No.62605/3787;使用Nonlinear USA Inc, Durham, NC USA)对FTSC/ iaf标记的蛋白点和考马斯色染色强度进行归一化。荧光点归一化为同一凝胶的蛋白质强度。
所有实验均为三次重复。在原始图像校正和背景相减后,基于三维高斯分布,对二维凝胶图像进行地标对齐,使对应的点相互匹配。光点强度归一化。自动确定二维凝胶图像中蛋白点之间的差异。
手工从胶体考马斯氏染色的2-DE凝胶中剔除方差分析p < 0.05的斑点。蛋白提取和酶解使用Perkin Elmer-Janus自动化工作站。消化后,样品在0.1%甲酸中重构,并使用Dionex U3000液相色谱系统(Dionex, Sunnyvale, CA, USA)和Daltonics HCT离子阱质谱仪(Bruker, Glasgow, UK)进行分析。在数据依赖的AutoMS(2)模式下获得肽片段的质谱,扫描范围为300-1500 m/z,从MS扫描(100-2200 m/z)中选择多达三个前体离子。前驱体在1.0 min的窗口内被主动排除,所有的单电荷离子都被排除。根据LC-MS /MS建立吉祥物通用文件,并使用Matrix Science webserver (www.matrixscience.com)检索吉祥物数据库NCBInr。默认的搜索参数是:酶=胰蛋白酶;最大缺失解理数=1;固定修饰=氨基甲酰(C);变量修饰=氧化(M);肽耐量±1.5 Da;MS/MS公差±0.5 Da;肽电荷=2 +和3 +。如果MASCOT评分高于95%置信度(p < 0.05),并且至少有一个肽的评分高于95%置信度(p < 0.05),则考虑鉴定出的蛋白质。这项分析是在英国阿伯丁大学蛋白质组学核心设施进行的。来自这些实验的所有质谱蛋白质组学数据已通过MassIVE存储库(数据集标识符为PXD022342)存入ProteomeXchange Consortium。
大肠杆菌中表达的人Rho-GDI1/ARHGDIA蛋白氨基酸24-204来自Novusbio (NBP1-50861, Centennial, CO, USA NBP1-50861), SDS-PAGE纯度> 95%,提供于20 mM Tris-HCl缓冲液(pH 8.0), 1 mM DTT, 10%甘油,不含防腐剂。重组人Memo在大肠杆菌中表达,纯度> 95%,以20 mM Tris-HCl缓冲液提供,pH 8.0, 50%甘油,5 mM DTT, 300 mM NaCl, 2 mM EDTA来自antibody -online (ABIN2130536, Aachen, Germany)。Moor et al.(2020)测定并报道了本实验中使用的这批重组Memo蛋白的铜还原氧化活性。
首先,作为假定的金属辅助因子预加载步骤,将2μg Memo与含有或不含10μM CuCl2的RCM缓冲液孵育。在4°C下孵育30分钟后,用Slide-A-Lyzer?MINI透析设备,3.5 K MWCO (69550 Thermo Fisher Scientific)在RCM缓冲液中去除游离CuCl2,在4°C下孵育30分钟。蛋白单独与1000 ng Rho-GDI1孵育15 min;Rho-GDI1 1000 ng, Memo 250 ng;1000 ng Rho-GDI1与250 ng CuCl2预处理Memo;和1000 ng Rho-GDI1,以100μM过氧化氢作为阳性对照(H1009 Sigma-Aldrich),每个实验一份,分别标记Rho-GDI1的总半胱氨酸和氧化半胱氨酸含量。
总半胱氨酸含量标记用5 mM Tris(2-羧乙基)膦(TCEP) (646547 Sigma-Aldrich)还原样品。总半胱氨酸含量用串联质量标签(TMT) iodoTMTsixplex?(90102 Thermo Fisher Scientific)标记,然后用20 mM 1DTT (43816 Sigma-Aldrich)淬火。
氧化半胱氨酸含量标记用500 mM碘乙酰胺(I6125 Sigma-Aldrich)处理30 min,然后用20 mM DTT淬火,用5 mM TCEP还原。用iodoTMTsixplex?标记样品的氧化半胱氨酸含量。
样品用SDS-PAGE清洁试剂盒(10074304 GE Healthcare Life Sciences,通过赛默飞世尔科学公司分发)沉淀,SDS-PAGE分离,考马斯蓝染色,手工条带切除。用胰凝乳酶在50°C下消化5 h,用LC-MS /MS (PROXEON与QExactive HF质谱联用,Thermo Fisher Scientific)分析蛋白质,两次注射5 μl消化物。肽被捕获在μPrecolumn C18 PepMap100 (5 μm, 100 ?, 300 μm × 5mm, Thermo Fisher Scientific)上,并在C18柱(5 μm, 100 ?, 75 μm × 15 cm, C18)上反冲洗,以5%乙腈到40%水,0.1%甲酸的梯度20分钟,流速为350 nl/min。Full Scan方法设置分辨率为60000,自动增益控制(AGC)目标为1E06,最大离子注入时间为50 ms。前体离子破碎的数据依赖方法采用以下设置:分辨率15,000,AGC为1E05,最大离子时间为110 ms,质量窗口1.6 m/z,第一质量100 m/z,碰撞能量27,填充率为1%,未分配和1 +离子的电荷排除,肽匹配优先。使用Proteome Discoverer 2.4.0.305对光谱进行解释,chymotrypsin规则允许多达8个缺失的切割,使用Cys上的碳酰胺甲基化(+ 57.021 Da),二氧化(+ 31.990 Da),三氧化(+ 47.985 Da)和iodoTMT标记(+ 324.216 Da)的可变修饰,以及蛋白质N-Term上的氧化(+ 15.99 Da)和乙酰化(+ 42.011 Da)的可变修饰。母体和碎片的质量公差分别设置为10 ppm和0.02 Da。高度自信肽谱匹配的严格目标错误发现率设置为0.01。只有当鉴定出两个满足1% FDR标准的独特肽时,才接受蛋白质鉴定。该分析在伯尔尼大学蛋白质组学质谱核心设施进行。得到的特定肽修饰的强度峰通过总信号强度归一化,并显示为非转换z分数的热图,即减去行平均值,然后除以行标准差。重组蛋白分析的所有质谱蛋白质组学数据已通过PRIDE存储库(数据集标识符为PXD022382)存入ProteomeXchange Consortium。
为进行免疫印迹,细胞和组织的裂解物在RIPA缓冲液或NP-40缓冲液(150 mM NaCl, 50 mM HEPES pH7.4, 25 mM NaF, 5 mM EGTA, 1 mM EDTA, 1% Nonidet P-40, 2 M Na - orthovandate和1 mM DTT,提供蛋白酶抑制剂lepeptin 10 μg/L,抑肽素10 μg/L, PMSF 1 mM)中裂解并变性。用SDS-PAGE分离蛋白质,将其转移到nitroccellulose或PVDF上,用Ponceau S染色,在干燥的脱脂乳中阻断5%-TBST或牛血清白蛋白(A9647 Sigma-Aldrich)在TBST中阻断3%,然后与Memo(1:2000,内部生产(Haenzi et al. 2014)或1:1000,HPA042603 Sigma-Aldrich), pERK (1:1000, sc-7383 Santa Cruz, Dallas TX, USA), tERK (1:1000, sc-93 Santa Cruz), actin (1:2000, A2066 Sigma-Aldrich), Rho-GDI1 (1:50 00, HPA042603 Sigma-Aldrich),ABIN969501 antibody -online), Rac1 (1:500, ab33186 Abcam, Cambridge UK), RhoA (1:500, NBP2-22529 NovusBio)和Klotho (1:100, AF1819 R&D Systems)。膜用抗小鼠或抗兔的山萝卜过氧化物酶偶联二抗(Milian Analytica, Rheinfelden Switzerland或ImmunoResearch,通过LubioScience GmbH, z
洛桑大学医院检测血清电解质:总钙(NM-BAPTA法)、磷酸(磷酸钼酸盐法)、肌酐(改良jaff<s:1>法)。按照制造商的说明,使用ELISA (Kainos Japan CY-4000)分析完整的FGF23。根据制造商的说明,使用荧光蛋白酪氨酸磷酸酶活性测定试剂盒(#K829, BioVision Inc., Milpitas CA, USA)测量肾裂解物中的蛋白磷酸酪氨酸磷酸酶(PTP)活性。为了定量肾脏匀浆中的Rho-GTPase活性,根据制造商的说明使用比色Rac1和RhoA G-LISA激活检测试剂盒(BK128-S和BK124-S Cytoskeleton Inc., Denver CO, USA)。
冷冻肾脏在TissueLyser (Qiagen, Hombrechtikon, Switzerland)中用金属珠均质。RNA提取使用TRI试剂溶液(AM9738 Ambion, Austin, TX, USA),用于所有下游应用。
使用Primescript RT Reagent kit (RR037 TAKARA,志贺,日本)逆转录每半肾1 μg RNA。用2 μl cDNA进行实时荧光定量PCR检测基因mRNA的表达情况。实验采用SYBR Green (Applied Biosystems, Foster City, CA UA),在7500 Fast机器(Applied Biosystems)上进行。样品一式三份,每个基因的总容积为20 uL,用肌动蛋白或GAPDH归一化。得到了每次运行的熔化曲线。程序设置为:95°C 20秒,40个循环(95°C 3秒,60°C 30秒),以及熔化曲线阶段:95°C 15秒,60°C 1分钟,以1%的斜坡速度上升到95°C(15秒),60°C 15秒。采用delta-delta CT方法对数据进行分析。引物从Microsynth (Balgach, Switzerland)订购,序列见补充表1。
对于RNAseq,从小鼠肾中提取的RNA两两合并到4个实验条件(2种处理状态,2种基因型)的3个单独的池中。使用片段分析仪(Advanced Analytical Technologies Inc.)评估和验证mRNA的质量。质量验证后,使用TruSeq搁浅RNA样品准备试剂盒(20020596 Illumina, San Diego CA, USA),总RNA为1ug,构建测序文库。利用Illumina HiSeq2500平台和单端reads (1 × 100)对cDNA进行测序。所有测序数据可在国家生物技术信息中心获取,序列读取档案,登录:PRJNA672305。使用Cutadapt (v. 1.3, Martin 2011)对纯化后的reads进行裁剪,使用seq_crumb (v. 0.1.8)进行过滤,并使用STAR (v. 2.4.0f1)对Mus musus (Ensembl version GRCh38.82)转录组进行比对。采用edgeR的修剪m值均值(trim Means of M-Values, TMM)方法对数据进行归一化,并采用生物导体封装极限的空间方法对数据进行变换。当没有一个样本的计数超过1 / 100万次读取时,低水平表达的基因被移除。只有标记为“蛋白质编码”的基因被保留用于统计检验。使用r4.1.0软件包gplots中的heatmap2函数构建热图,使用对数转换归一化读的z分数。
参数连续变量的分析采用t检验或方差分析,所有实验组的多重检验采用Bonferroni校正后验。对非参数连续变量的分析采用Kruskal Wallis检验,然后对所有实验组的多重检验进行Dunn校正。采用GraphPad Prism 5.0进行分析,双尾p值< 0.05为显著性。
使用limma和DESeq2 (R版本为3.2.1,limma版本为3.22.7,DESeq2版本为1.8.1)对RNAseq数据进行分析。由于实验采用2 × 2因子设计,采用两种小鼠基因型和两种处理状态,因此以所有4组为因素建立线性模型。随后,使用缓和t检验对以下组提取感兴趣的对比:“KOFGF-KOV”(备忘录cKO中的治疗效果);2 .“WTFGF-WTV”(在固定对照基因型中的处理效果);3 .“KOFGF-WTFGF”(在fgf23处理小鼠中的KO基因型效应);“KOV-WTV”(KO基因型效应);“(KOFGF-KOV)-(WTFGF-WTV)”表示基因型与治疗之间的相互作用。通过Benjamini-Hochberg方法调整p值,控制所有5个对比的错误发现率(FDR),作为多个测试条件的校正。所有测序数据可在国家生物技术信息中心测序读取档案中获得,识别码为PRJNA672305。使用2020年11月17日访问的大猩猩(Eden et al. 2009)对基因本体的富集进行评估,使用按调整p值排序的全基因列表。
本研究得到了瑞士沃州和伯尔尼兽医服务部门的批准。
摘要
介绍
材料与方法
结果
讨论
缩写
参考文献
致谢
作者信息
道德声明
补充信息
搜索
导航
#####
在目前的研究中,我们开始测试fgf23驱动的信号传导是否不仅在培养细胞中依赖于Memo,而且在更复杂的啮齿动物领域中也依赖于Memo。为此,我们产生了可诱导的全身Memo cKO和不含Cre转基因的固定对照小鼠,并按照前面的描述在C57BL/6背景下维持它们(Moor et al. 2018)。我们用他莫昔芬对两种基因型小鼠进行全身切除Memo1外显子2。如图1A所示,我们将他们随机分为腹腔注射FGF23或载体注射。注射FGF23可使循环中完整的FGF23增加14倍(补充图1A)。先前的研究发现,在培养的memo缺陷细胞中,几种信号反应存在缺陷,包括ERK信号(Frei et al. 2016;Schotanus and Van Otterloo 2020)。我们评估了全肾溶物中磷酸化和总ERK蛋白的水平,并检测到FGF23处理的对照小鼠中磷酸化ERK的增加,而FGF23处理的Memo cKO动物中磷酸化ERK的增加(图1B, C)。接下来,我们分析了基因型组和处理组小鼠肾脏中ERK下游基因的表达。FGF23处理1小时后,仅在对照小鼠中持续增加Cyp24a1(图1D)和Egr1表达(图1E),而在FGF23处理的cKO中没有增加。这些数据强调了Memo在促进肾脏中fgf23驱动的细胞信号传导中的作用。
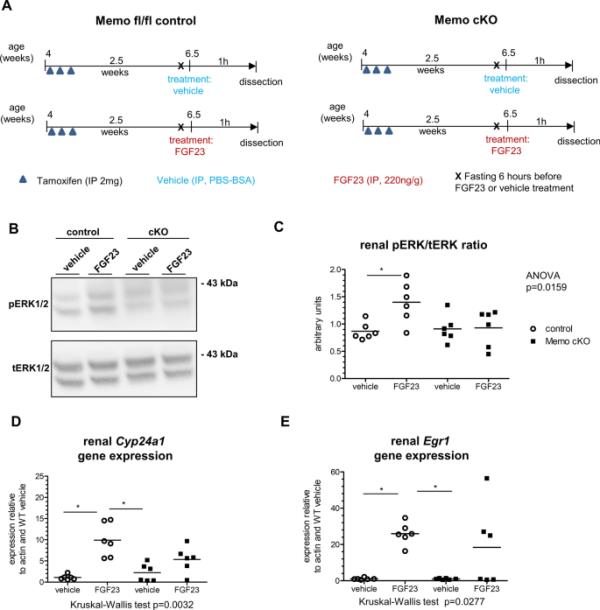
fgf23诱导的全身Memo KO (cKO)小鼠肾脏信号通路受损。在Memo消融2.5周后,将小鼠随机分为重组FGF23或载药腹腔注射(A)。在FGF23或载药后1小时,通过Western blot评估磷酸化(p)与总(t)肾ERK蛋白(B)。密度定量显示,Memo cKO肾脏中,FGF23对pERK/tERK比率的反应减弱(C)。通过qPCR评估Cyp24a1 (D)和egr1 (E)的靶基因反应。各组间采用Bonferroni后验(C)或Kruskal-Wallis后验(D, E)进行方差分析。P *,后验P < 0.05。每种情况n=6 (B-E)
这些发现没有受到Memo cKO动物严重表型的影响:本研究中纳入的小鼠都是在先前报道的早衰迹象出现之前进行研究的(Haenzi et al. 2014),并且仍然健康,如体重(补充图1B)、血清肌酐(补充图1C)、钙和磷酸盐浓度(补充图2D, E)所示,这些基因型之间总体上是可比较的。此外,我们确保Memo的KO在所有分析样品中都是成功的。(补充图1F)。总体而言,小鼠模型如预期的那样显示Memo耗竭,但观察到的信号表型不是由该模型后期发展的疾病特征引起的。
FGFR信号由负调节因子控制。因此,我们通过qPCR测量了Memo cKO肾脏中由Il17rd编码的SEF,并用FGF23或载体刺激对照,发现Il17rd转录本在肾脏中的治疗和基因型中保持不变(补充图1G)。我们之前的研究表明,Memo cKO动物在重组后12-14周或5周发生肾衰竭,在晚期CKD中,Klotho蛋白的严重丢失加重了肾脏排泄磷酸盐的能力(Koh等人,2001;Moor et al. 2018)。因此,我们在本研究中检测了重组后6.5周或2.5周的对照组和KO动物的Klotho蛋白水平。FGF23共受体Klotho的肾蛋白量在不同实验组之间没有变化(补充图1H)。总之,共受体Klotho或即时负反馈调节因子的丰度没有改变,因此不太可能是在Memo cKO动物中观察到的肾脏FGF23信号表型的原因。
为了进一步了解在Memo cKO中受到干扰的fgf23依赖性信号通路,我们评估了fgf23处理和载体处理的Memo cKO小鼠和对照小鼠成对汇总的肾脏样本的转录组(图2A,补充表2-6)。在FGF23治疗1小时后的急性反应中,我们在非监督聚类(补充图2A)、主成分分析(补充图2B)和对数转换归一化reads的热图分析(图2B)中发现了一致的基因型效应,但对整体转录组没有很强的治疗效应。然而,对照基因型中FGF23增加了13个转录本,Memo cKO中增加了1个转录本(图2C,补充图2C)。在13个fgf23依赖性增加的转录本中,4个编码双特异性磷酸酶(DUSP)。因此,在对照小鼠中,FGF23处理后,具有功能注释为丝裂原活化蛋白激酶(MAPK或ERK)磷酸酶活性的基因本体转录本被强烈富集(补充图3,补充表3)。相比之下,FGF23在Memo cKO中处理后,与MAPK磷酸酶活性相关的富集程度较低(补充图4)。补充表2)。在基因型和FGF23治疗的相互作用分析中,这种效应仍然显著(补充图5,补充表6),表明在全球转录组水平上,Memo控制对FGF23的反应更强。对所选转录本的进一步分析表明,FGF23驱动的Cyp24a1、Cyp27b1或Hbegf的变化受损,以及Memo cKO中Memo1转录本水平持续降低(图2C, D)。总体而言,我们发现FGF23对肾脏转录的基因型不同,涉及与MAPK磷酸酶活性相关的转录本。
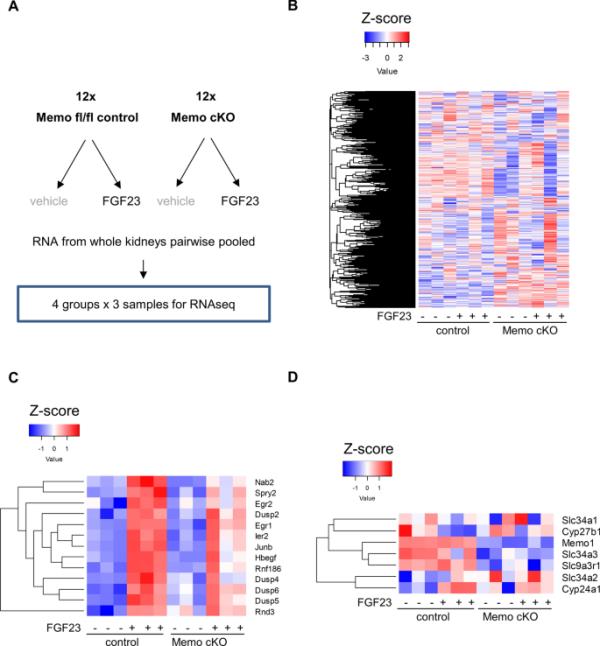
肾脏RNAseq分析显示Memo cKO和对照组之间存在全局转录组学差异。实验条件和设置如图(A)所示,每个测序文库都有两只动物。全球转录组热图显示了Memo cKO和对照组之间微妙的基因型差异(B)。所有实验组中FGF23和载体之间13个不同表达的转录本(c)和选定的编码FGF23信号传导蛋白的转录本(D)
接下来,我们旨在确定氧化还原蛋白Memo是否通过PTPs影响肾脏FGF23信号传导。氧化还原蛋白Memo是诱导NOX活性所必需的(MacDonald et al. 2014)。在细胞信号传导过程中需要nox来调节PTPs (Lee et al. 2007), PTPs组成性地控制激酶级联反应,如ERK家族。ptp受细胞特定空间区域的半胱氨酸氧化调节(Miki和Funato 2012;Truong和Carroll 2012),并显示磷酸酶活性在低剂量氧化剂的作用下增加,但在高剂量氧化剂的作用下减少(Wright et al. 2009)。
由于这些原因,我们旨在通过调节PTP活性来破译fgf23驱动的细胞信号传导中Memo氧化还原功能的下游效应。在用FGF23或对照剂治疗1小时之前,我们先用全局PTP抑制剂原钒酸钠治疗Memo cKO和对照小鼠1小时。在PTP抑制剂处理的动物中,我们发现与对照NaCl 0.9%处理的动物(补充图6A,左图)相比,原钒酸钠处理的动物全肾PTP活性降低(右图)。然而,在原钒酸盐部分抑制PTP后,我们发现FGF23处理在对照动物肾脏中诱导的PTP活性相对增加。(补充图6A,右面板)。这种效果在Memo cKO小鼠中减弱了。在本实验环境下,Memo cKO小鼠中Memo1的表达持续减少,证实了这些实验动物中重组的有效性(补充图6B)。综上所述,FGF23对肾脏PTP活性的影响是基因型依赖的,在对照基因型动物中,FGF23对PTP活性的影响更强,变化更少,但只有在通过药物抑制PTP时才可见。
我们假设在fgf23驱动的细胞信号传导过程中,Memo的氧化还原功能可能会扰乱细胞内氧化还原稳态。为了验证这一点,我们使用氧化还原蛋白质组学和氧化敏感荧光探针来确定作为氧化反应中间体的硫醇和羰基的全球丰度,这些中间体是FGF23或经载体处理的Memo cKO和对照组肾脏中的氧化反应。通过IAF评估的细胞总蛋白硫醇水平在处理过的对照组和Memo cKO小鼠之间具有可比性(图3A)。然而,在fgf23处理的Memo cKO中,IAF信号增加到比对照组更高的水平,分别为2.54倍和2.17倍。这些结果表明,fgf23驱动的整体半胱氨酸氧化在基因型之间存在显著差异(图3A)。在FGF23处理的对照组中,总蛋白羰基含量增加了1.95倍,但在Memo cKO中没有显著变化(图3B),这表明Memo cKO肾脏异常处理活性中间体(例如通过过氧化导致羰基),导致Memo cKO肾脏中过量的蛋白质硫代化。总的来说,我们观察到在fgf23驱动的信号传导过程中,细胞内氧化还原状态的明显基因型依赖的动态变化。
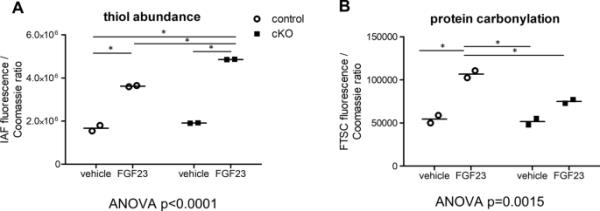
fgf23驱动的肾脏整体氧化还原蛋白质组的变化。对氧化还原敏感的硫醇偶联物5-碘乙酰氨基荧光素(IAF)的定量分析显示,在FGF23的作用下,对照小鼠的蛋白质硫醇含量适度增加了2.17倍,但Memo cKO的蛋白质硫醇含量增加了2.54倍(a)。但Memo cKO仅为1.45倍(B)。每个数据点代表2只动物合并肾半的3个技术重复的平均值(每种情况n=2 × 2)。测量的强度通过考马斯氏染色总蛋白归一化。各列数据采用Bonferroni后验方差分析,各组间显著后验为(*)。FGF23,成纤维细胞生长因子23
为了确定氧化态改变的主要蛋白质的分子特性,我们使用2DGE重新分析了图3A, B所示的肾脏样品,并通过等电点和质量分离。随后,我们使用软件辅助测定改变的蛋白点。补充图7显示了对照动物蛋白点的2DGE分析,其中comasmase, IAF或FTSC信号在基因型之间存在差异。表1为MALDI-TOF/MS测定的相应蛋白特性。我们在Memo cKO中发现谷胱甘肽(GSH)合成酶硫醇信号减弱,硒结合蛋白1升高。除了进一步与细胞骨架和碳水化合物代谢相关的差异氧化蛋白外,我们还发现了一种细胞信号相关蛋白,Rho-GDP解离抑制剂1 (Rho-GDI1),一种Rho-GTPases伴侣蛋白。与对照组相比,fgf23治疗的Memo cKO中Rho-GDI1的肾硫醇含量增加了1.69倍(表1第378点)。总之,一项对蛋白质组的氧化还原敏感研究显示,在Memo cKO和对照组之间,Rho-GTPase相互作用蛋白Rho-GDI1的半胱氨酸氧化状态发生了改变。
接下来,我们的目标是确定这一发现的生化意义。我们实验测试了Memo和Rho-GDI1在功能上相互作用的可能性。为此,我们分析了重组Memo是否可以改变Rho-GDI1中唯一的半胱氨酸——半胱氨酸79的氧化状态,从而可能影响其功能。为此,我们量化了Rho-GDI1 Cys79在天然肽中的翻译后修饰,并在无细胞环境中使用巯基特异性iodoTMT(图4A)。Memo预先加载CuCl2,假定的辅助因子铜,然后透析去除游离铜,如前所述(16)。我们发现Rho-GDI1的Cys79可逆氧化为亚磺酸盐可以忽略不计,但在Rho-GDI1的Cys79处检测到不可逆的二氧化为亚砜基(SO2H)和一定程度的三氧化为磺酰基(SO3H)(图4B)。H2O2作为不可逆氧化的阳性对照。这些结果表明,Memo在体外直接氧化Rho-GDI1。

重组Memo与Rho-GDI1在无细胞条件下的氧化相互作用。A为重组Rho-GDI1中Cys79修饰分析的实验流程,分别用或不加铜预载Memo或H2O2孵育。简而言之,不可逆二氧化(SO2H)和三氧化(SO3H)通过MALDI-TOF/MS直接检测,可逆单氧化(SOH)肽通过碘乙酰胺(IAA)阻断可达半胱氨酸后,使用iodoTMT串联质谱标签(TMT)检测;在没有IAA阻断的情况下检测总可达半胱氨酸。RhoGDI1与H2O2孵育作为阳性对照引起三氧化(B,第一排),与铜预载和透析的Memo孵育引起RhoGDI1 Cys79的二氧化和较小程度的三氧化(B,蓝色矩形)。Memo或铜预加载Memo的添加不会改变TMT标签强度(B,第三行和蓝色矩形)。只有一个样品有一些剩余的游离硫醇(B,第4行),并且在所有IAA阻断的样品中,存在IAA烷基化(B,第5行)。B中的信号强度被归一化为检测到的肽总数强度,并显示为原始未转换数据的z分数。或Kruskal-Wallis检验,Dunn多重检验校正(B),后验p < 0.05。每个条件N=3个独立实验
接下来,我们的目标是描述Rho-GDI1是否在fgf23驱动信号的早期信号级联中是直接需要的,或者它是否通过影响其他介质在这一过程中发挥不太直接的作用。我们在稳定表达Klotho (HEK293- kl)的HEK293细胞系统中对Rho-GDI1进行了遗传操作(补充图8)(Diener et al. 2015)。Rho-GDI1在HEK293细胞中内源性表达(Boulter et al. 2010)。使用两种不同的sirna靶向Rho-GDI1,我们将Rho-GDI1敲低至1%或30%的模拟转染细胞(补充图9A, B和10A, B)。接下来,我们在两个不同的时间点评估细胞对FGF23的信号反应。在Rho-GDI1敲低和对照细胞之间,FGF23刺激下ERK磷酸化没有差异(补充图9C, D和10C, D)。相反,我们测试了Rho-GDI1丰度的增加是否会影响FGF23信号传导。我们短暂过表达Rho-GDI1(补充图11A)。同样,Rho-GDI1过表达和模拟转染细胞对FGF23刺激的ERK磷酸化没有差异(补充图11B, C)。总体而言,HEK293-KL细胞中Rho-GDI1的敲低或过表达不会改变FGF23刺激下ERK磷酸化水平。
Rho-GDI1是一种伴侣蛋白和小rho - gtpase(如RhoA和Rac1)的调节剂,这两种蛋白对包括FGFR信号在内的细胞信号传导特别重要(13)。通过半胱氨酸氧化促进RTK信号,一氧化氮介导的ptp失活需要Rac1 (Wright et al. 2009;Miki and Funato 2012;Truong and Carroll 2012)。我们在图4中表明,Memo直接氧化Rho-GDI1。而且,Memo可以直接氧化RhoA (MacDonald et al. 2014)。接下来,我们研究了在Memo cKO肾脏中Rho-GDI1、Rac1和RhoA的总蛋白水平是否改变。
Rho-GDI1丰度是Rho-GTPase活性的重要调节因子(Boulter et al. 2010)。因此,我们检测了Memo cKO和对照动物肾脏中Rho-GDI1的总体丰度。Rho-GDI1丰度在基因型和FGF23处理状态之间具有可比性(图5A,定量见图5E)。在Rho-GDI1总量不变的情况下,fgf23处理的对照小鼠中Rho-GDI1硫醇含量的降低与Rho-GDI1 Cys79的短暂氧化相对应,从而释放并激活结合的Rho-GTPase。此前对RhoA-Rho-GDI1复合体也有类似的观察结果(Kim et al. 2017)。备忘录要求增长要素驱动RhoA relocalization的膜(约尔et al . 2008年),和引人注目的是,RhoA蛋白质丰度和活动增加肾脏的备忘录cKO老鼠在车辆和FGF23治疗组(图5 d, E, F)。丰富的Rho-GTPase Rac1在所有条件不变(图5度,量化在图5 E),然而,FGF23 Rac1活动倾向于增加10%在整个肾脏FGF23治疗控制老鼠(图5),类似于在细胞培养中对FGF2处理的Rac1活性的反应(Shin et al. 2006;Barrios and Wieder, 2009)。重要的是,在FGF23处理的Memo cKO动物中,没有这种增加Rac1活性的趋势。肾Memo蛋白在Memo cKO中如预期的那样减少(图5D, E)。总之,除了Rho-GDI1硫醇信号增加外,我们发现由于Memo1功能丧失,肾Rho-GDI1相互作用蛋白RhoA的丰富度和活性显著增加。
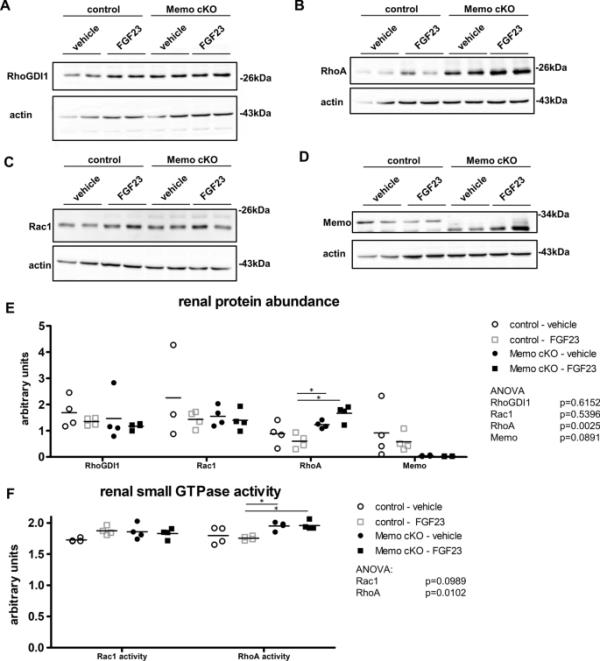
Memo cKO (cKO)小鼠肾脏Rho-GTPase网络功能异常。整个肾脏的Western blots显示了指定的蛋白质(a - d)和它们各自的密度定量对抗肌动蛋白作为负载控制。采用G-LISA法测定肾脏Rac1和RhoA活性(F)。E-F数据采用Bonferroni校正方差分析(各6个后验)。*后验p < 0.05。A-F中每个条件n=4。每个条件(A-F) n=4;除E中n=3外(Rac1:对照组)
下载原文档:https://link.springer.com/content/pdf/10.1007/s12079-022-00710-1.pdf
 微信扫一扫打赏
微信扫一扫打赏

